آموزش ساخت ورودی برای نرم افزار محاسباتی گوسین
- توضیحات
- منتشر شده در شنبه, 05 مهر 1393 14:41
- نوشته شده توسط حسین حجی آبادی
- بازدید: 36752
نرم افزارهای محاسبات علمی به رغم اینکه بسیار پیچیده و متشکل از هزاران خط کد هستند، ظاهر بسیار ساده ای دارند. برخلاف بسیاری از نرم افزارهای متداول مانند نرم افزارهای آفیس، نرم افزارهای طراحی، فتوشاپ و غیره که از تعداد زیادی منو و دکمه تشکیل شده اند، نرم افزارهای محاسباتی فاقد رابط گرافیکی اند (و یا دارای رابط گرافیکی بسیار ساده در برخی از پلت فرمها هستند). تنظیمات و اطلاعات مورد نیاز این نرم افزارها نه از طریق رابط گرافیکی بلکه به وسیله یک فایل ساده متنی به این نرم افزارها منتقل می شود.به صورت کلی انجام یک محاسبه علمی را می توان به سه مرحله تقسیم کرد: 1) ساخت فایل ورودی 2) اجرای دستور محاسبه 3) تفسیر فایل خروجی.فایل ورودی برای هر نرم افزاری دارای قالب (فرمت) خاص خود است و از کلمات کلیدی خاصی تشکیل شده است که به نرم افزار می گوید: برای چه ساختاری، چه محاسبه ای، و به چه نحوی انجام دهد.
در این مقاله و چند مقاله آتی قالب فایل ورودی نرم افزارهای محاسباتی مختلف معرفی خواهد شد. در اولین مقاله از این سری از مقالات ابتدا به سراغ متداول ترین نرم افزار محاسباتی (Gaussian 09)خواهیم رفت.
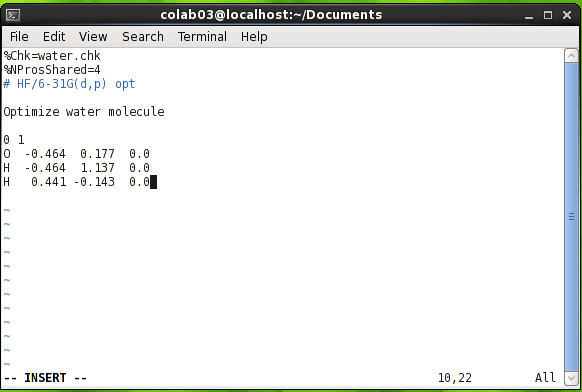
همانطور که گفته شده فایل ورودی نرم افزار Gaussian یک فایل ساده متنی (با استانداردASCII) است. ساده بودن این فایل به این معنی است فایل باید فاقد هرگونه قالب نگارشی مانند نوع و اندازه قلم، پاراگراف بندی، رنگ و غیره باشد. به همین علت برای تهیه فایلهای ورودی باید از یک نرم افزار text editor ساده مانند vi یا gedit در محیط یونیکس و یا notepad در محیط ویندوز استفاده شود.
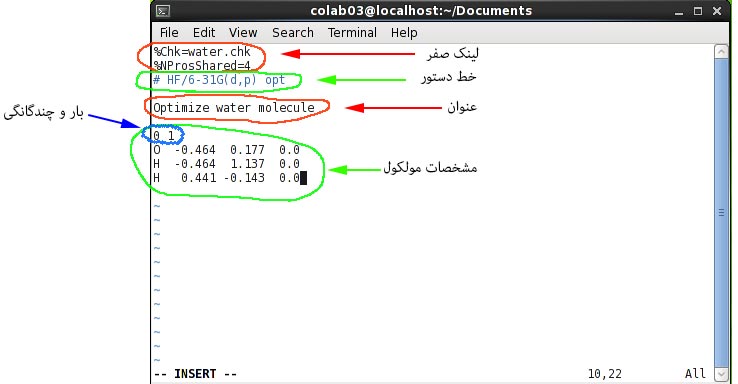
برای آشنایی بهتر با فایل ورودی Gaussian یک نمونه فایل ورودی در شکل بالا آورده شده است. همانطور که در این شکل ملاحظه می شود، فایل ورودی از قسمت های مختلفی تشکیل شده که در ادامه هر یک از قسمت ها توضیح داده خواهد شد. توجه کنید که برخی از این قسمت ها به یک خط خالی ختم می شوند و برخی دیگر نیازی به خط خالی در انتها ندارند که این خط خالی قسمتی از قالب ورودی است و دقیقا باید رعایت شود. در ورودی نمایش داده شده اولین قسمت link 0 نام دارد که با علامت % شروع می شود. لینک صفر حاوی کلید واژه هایی است که نحوه ی به کار گیری منابع سخت افزاری مانند آدرس و نام فایل های اسکرچ، میزان حافظه ای که باید استفاده شود، تعداد هسته سی پی یو و غیره را مشخص می کنند. یک فایل ورودی می توانند بدون لینک صفر و یا دارای بیش از یک لینک صفر باشد. در مثال بالا عبارت %Chk=water.chk مشخص می کند که نتایج محاسبات باید در فایلی با نام water.chk ذخیره شود و عبارت %nProsShared=4 تعیین می کند که باید از چهار هسته پردازنده برای انجام محاسبه استفاده شود. توجه کنید که لینک صفر به خط خالی ختم نمی شود.
قسمت بعدی در فایل ورودی خط دستور یا route section است. خط دستور همواره با علامت # شروع می شود. این خط به نرم افزار می گوید که چه نوع محاسبه ای را انجام دهد و همچنین شامل تنظیمات نرم افزاری مربوط به انجام محاسبه است. برای کنترل سطح ذخیره سازی اطلاعات در فایل خروجی می توان به جای علامت # از علامت #T و یا #P استفاده کرد که به ترتیب باعث خلاصه شدن و افزوده شدن اطلاعات این فایل می شوند. در فایل ورودی بالا سه کلید واژه در خط دستور وجود دارد. کلید واژه اول عبارت HF است که مشخص می کند محاسبه باید با استفاده از روش هارتری- فک انجام شود. کلید واژه دوم عبارت 6-31G(d,p) است که نوع تابع پایه مورد استفاده را تعیین می کند. دقت نتایج یک محاسبه ساختار الکترونی وابسته به تابع پایه آن است. هرچه تابع پایه مورد استفاده بزرگتر باشد، دقت نتایج نیز بیشتر است. کلید واژه سوم عبارت opt است که نوع محاسبه را تعیین می کند. در این مثال یک محاسبه بهینه سازی ساختار (optimization) از نرم افزار درخواست شده است. توجه کنید که ترتیب کلید واژه ها در خط دستور اهمیت ندارد. این کلید واژه ها را می توان با استفاده از فضای خالی، کاما و یا اسلش (space, commas or slash) از یکدیگر جدا کرد. همچنین ورودی نرم افزار به صورت کلی حساس به حروف بزرگ و کوچک نیست بنابراین خط دستور بالا را می توان به شکل های زیر نیز نوشت:
# hf 6-311G(d,p) OPT
# HF,OPT,6-311G(d,P)
# opt 6-311g(d,p)/hf
در پایان توجه کنید که خط دستور باید به یک خط خالی ختم شود.
قسمت بعدی در فایل ورودی، عنوان(title) کار است. عنوان یک کار معمولاً شامل توصیفی مختصر از کار است. عبارات نوشته شده در این قسمت توسط نرم افزار تفسیر نمی شود و می تواند شامل هر عبارت دلخواهی باشد با این حال عنوان نباید شامل کارکترهای @ # ! - _ \ و کارکترهای کنترل (به خصوص Ctrl + G) باشد. توجه کنید که عنوان باید به یک خط خالی ختم شود.
آخرین قسمت در ورودی بالا شامل مشخصات مولکول مورد نظر است. در این قسمت ابتدا به ترتیب بار و چندگانگی (multiplicity) مولکول مورد نظر و سپس مختصات آن نوشته شده است. براین اساس مشخص است که مولکول مثال ما خنثی و یکتایی (singlet) است. همچنین با نگاه کردن به مختصات مولکول می توان دریافت که محاسبه بر روی یک مولکول آب انجام خواهد شد. در مثال ما ساختار مولکول آب با استفاده از سیستم مختصات کارتزین (دکارتی) نوشته شده است. مزیت استفاده از سیستم از کارتزین عمومی بودن آن است. به این معنی که این سیستم ساختار یکسانی در تمام نرم افزارهای محاسباتی و رسم ساختار مولکولی دارد. با این حال مختصات دکارتی تنها سیستم مختصات مورد قبول نرم افزار Gaussian نیست. هر چند ساختار یک مولکول را می توان به صورت دستی در فایل ورودی نوشت ولی این کار بجز برای ساختارهای بسیار کوچک بسیار طاقت فرسا است و معمولاً برای تهیه مختصات مولکول از یک نرم افزار سه بعدی رسم ساختار مولکول مانند Gaussview استفاده می شود.
در پایان ذکر چند نکته درباره فایل ورودی Gaussian ضروری است.
- با استفاده از علامت ! می توان در هر قسمت از فایل ورودی توضیح (comment) نوشت.
- برای وارد کردن محتویات یک فایل در فایل ورودی می توان از علامت @ به شکل @filename استفاده کرد. با این کار نرم افزار Gaussian محتویات فایل خارجی را قرائت و به جای این عبارت قرار خواهد داد.
- برخی از کلید واژه های Gaussian دارای آپشن های خاصی هستند که این آپشن ها را می توان به یکی از شکل های زیر وارد کرد.
Keyword=option
Keyword(option)
Keyword=(option1, option2, …)
Keyword(option1, option2, …)
به عنوان مثال برای تعیین حداکثر تعداد سیکل های بهینه سازی، کلید واژه opt را می توان همراه با آپشن maxcycle به صورت: opt(maxcycle=n) به کار برد که n برابر با تعداد حداکثر سیکل های مورد نظر است.
- کلید واژه ها و آپشن های Gaussian می توانند به کوتاه ترین شکل بی مانندشان خلاصه شوند. به عنوان مثال آپشن conventional ( مربوط به کلید واژه SCF) می تواند به صورت conven نوشته شود. ولی این آپشن را نمی توان به صورت conv خلاصه کرد زیرا این عبارت حروف اول آپشن convergence نیز است.
- تمامی پارامترهای ورودی Gaussian دارای مقادیر پیش فرضی هستند که در صورت مشخص نشدن آنها در فایل ورودی از مقادیر پیش فرض استفاده می شود. به عنوان مثال در صورت مشخص نشدن نوع محاسبه در فایل ورودی، یک محاسبه انرژی (SP) انجام خواهد شد.
دیدگاهها
سلام و اوقات شما هم خوش
دقیقا کدوم اعداد رو با هم مقایسه کردید؟
اعداد خروجی گوسین با چیزی که در گوسویو مشاهده میکنید باید یکسان باشند! البته گوسویو انرژی اوربیتالها رو یک رقم رند میکنه! اختلاف اعدادی که میگید در چه حدی است؟
با احترام
مسیله ای که باش برخورد کردم و برام جای سوال داشت را با اجازتون مطرح میکنم اگر اطلاع داشتید و امکانش بود از راهنماییتون استفاده کنم
در مورد نمایش اوربیتالها توسط گاوس ویو- وقتی فایل خروجی گاوسین را باز میکنم سطوح انرژی با مقادیر مشخص نشان داده میشود اما با لود کردن فایل chk (جهت نمایش حجیم حالات اوربیتالی) سطوح انرژی (مقدارشان) عوض میشود- مسیله چیست؟ درک نمیکنم
و متوجه نمیشم-درست و غلط بودن کدام حالت؟!
ممنون از سایت خوب و ایجاد فرصتی برای به اشتراک گذاشتن تجربیات علمی - تحقیقاتی
عاقبت همگی خوبان خوش ان شالله
مسیله ای که باش برخورد کردم و برام جای سوال داشت را با اجازتون مطرح میکنم اگر اطلاع داشتید و امکانش بود از راهنماییتون استفاده کنم
در مورد نمایش اوربیتالها توسط گاوس ویو- وقتی فایل خروجی گاوسین را باز میکنم سطوح انرژی با مقادیر مشخص نشان داده میشود اما با لود کردن فایل chk (جهت نمایش حجیم حالات اوربیتالی) سطوح انرژی (مقدارشان) عوض میشود- مسیله چیست؟ درک نمیکنم
و متوجه نمیشم-درست و غلط بودن کدام حالت؟!
ممنون از سایت خوب و ایجاد فرصتی برای به اشتراک گذاشتن تجربیات علمی - تحقیقاتی
عاقبت همگی خوبان خوش ان شالله
مسیله ای که باش برخورد کردم و برام جای سوال داشت را با اجازتون مطرح میکنم اگر اطلاع داشتید و امکانش بود از راهنماییتون استفاده کنم
در مورد نمایش اوربیتالها توسط گاوس ویو- وقتی فایل خروجی گاوسین را باز میکنم سطوح انرژی با مقادیر مشخص نشان داده میشود اما با لود کردن فایل chk (جهت نمایش حجیم حالات اوربیتالی) سطوح انرژی (مقدارشان) عوض میشود- مسیله چیست؟ درک نمیکنم
و متوجه نمیشم-درست و غلط بودن کدام حالت؟!
ممنون از سایت خوب و ایجاد فرصتی برای به اشتراک گذاشتن تجربیات علمی - تحقیقاتی
عاقبت همگی خوبان خوش ان شالله
سلام
دستور rwf فضای مورد استفاده برای فایلها اسکرچ که روی دیسک ذخیره میشه رو تغییر میده. %rwf=آدرس و اسم فایل
برای تعیین کردن مقدار حافظ اصلی مورد استفاده از این عبارت استفاده کنید. %mem= مقدار حافظه
هردوی این عبارات باید قبل از خط دستور قرار بگیره
با احترام
سلام
کیورد scrf برای محاسبات در محیط محلوله و اگر منظورتون همین بوده، بله باید برای محاسبه فرکانس هم باید باشه. البته وقتی از خروجی ورودی میسازید معمولا کلمات کلیدی هم منتقل میشه ولی حتما چک کنید که باشه.
با احترام
ببخشید برای محاسبه دانسیته که گفتین pop=reg داخل خروجی باید چی سرچ کنم تا دانسیته رو که حساب کرد ببینم....
ممنونم از پاسخگویتون
سلام
تا جایی که من میدونم گوسین رفرکتیو ایندکس رو حداقل به صورت مستقیم محاسبه نمیکنه! برای بدست آوردن این پارامتر احتمالا باید محاسبات دیگه ای انجام بدید و بعد از نتایج اونها رفرکتیو ایندکس رو حساب کنید.
برای محاسبه دانسیته الکترونی هم از pop=reg استفاده کنید.
با احترام
دستوری که برای بدست اوردن رفرکتیو ایندکس یا دانسیته هستش رو می تونید لطف کنید بهم بگین